Molecular Self-Assembly
Controlling their structure and dynamics
There is a certain sense of wonder when inanimate objects spontaneously arrange themselves to form an ordered structure… In fact, cells are constantly rely on self-assembly processes during their live and replication – with the perhaps the most known example being the self assembly of the phospholipids which compose their membranes. The process by which molecules spontaneously adopt a defined arrangement, i.e. without any external action, is known as molecular self-assembly and its a core concept in supramolecular chemistry. This process offers a myriad of opportunities and is in key in a bottom-up design of nanoarchitectures capable of meeting specific needs (e.g. photo-voltaic devices, CO2 capturing materials, … ). The spontaneous arrangement is often guided by weak reversible inter-molecular interactions such as hydrogen bonding, van-der-Waals forces, hydrophobic forces, among others. Our research efforts are focused in using molecular engineering to control these interactions and thus the resulting molecular structures its associated properties. Our background, put us in a privileged position do to so as all required tools are in place: possibility to simulate large structures (>10nm) over long periods of time (> 1µs), accurately describe the adsorption process with dedicated inter-atomic potentials built from first principles calculations and our broad expertise in surface science. Some of our major achievements are:
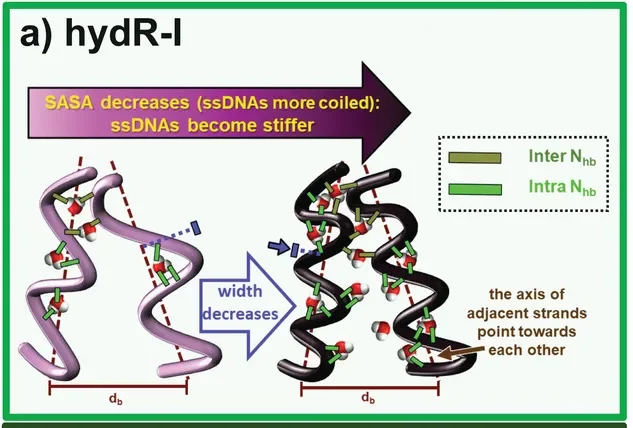
We provided the first atomically detailed understanding of long-range hydration-induced deformations in a biomolecular system. Controlling shape, size and stiffness through hydration is not novel – it is, in fact, a phenomenon ingeniously explored in Nature (being plants among the most familiar examples). However, beyond recognizing it, understanding such phenomena is key to harness it in emerging technologies. Our work elucidated the connection between picosecond hydrogen-bond dynamics and the cooperative deformation of single-stranded DNA brushes over hundreds of nanoseconds, essential for smart-responsive materials and label-free sequencing devices.
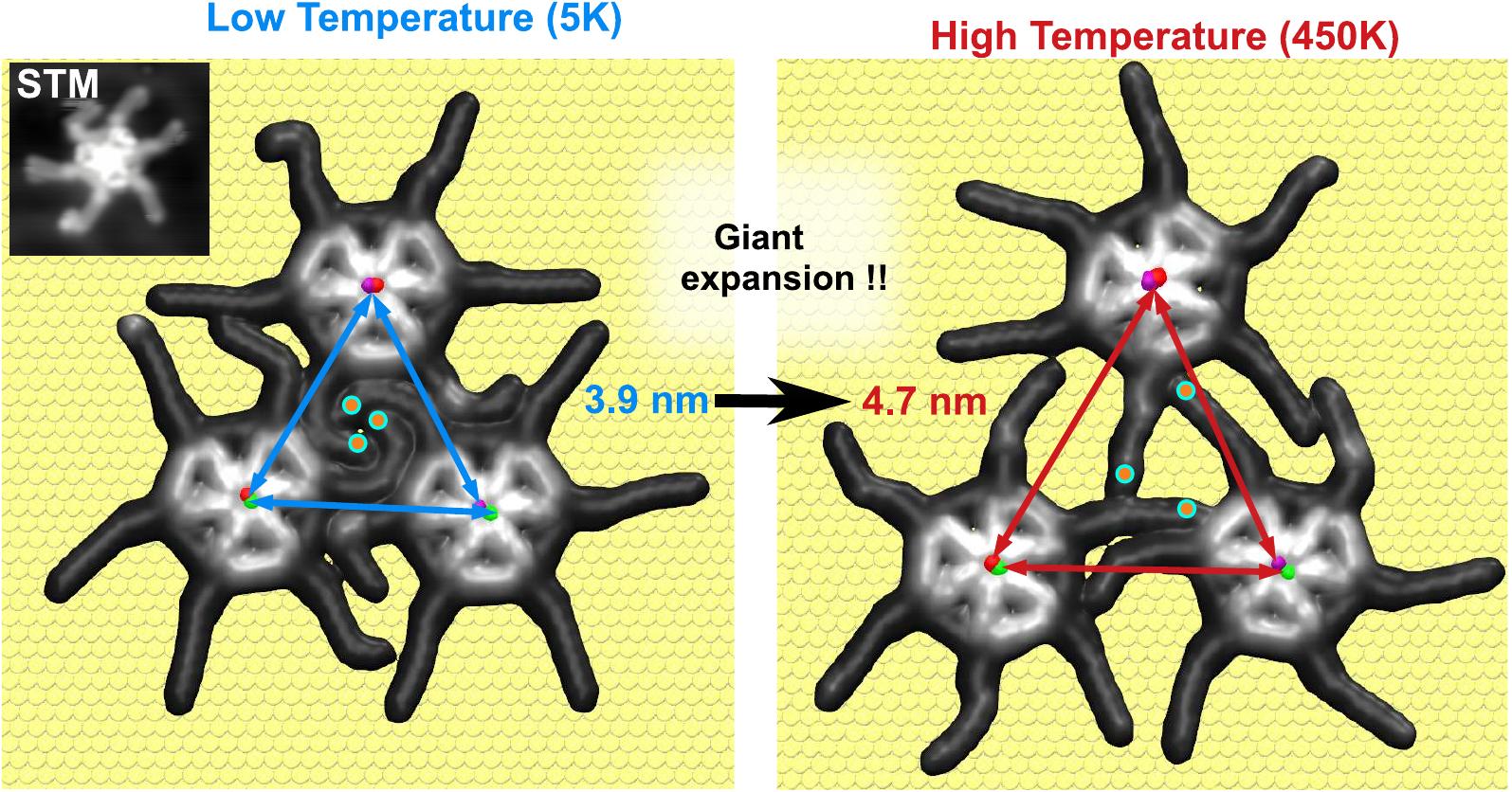
We discovered a novel method to control the thermal expansion coefficient (TEC) of molecular assemblies. By adding alkyl chains to a molecular core, we achieved a record high TEC of 980 ± 110 × 10⁻⁶ K⁻¹, over 70 times greater than that of gold (14 × 10⁻⁶ K⁻¹). This significant increase is attributed to the entropic fluctuations of the alkyl chains.
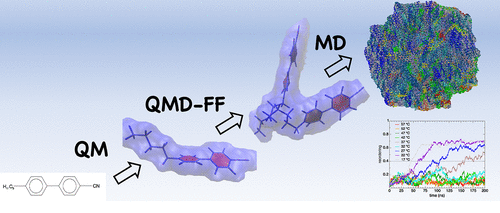
Together with the group of Prof. Giacomo Prampolini (in CNR Pisa) we demonstrated the predictive power of quantum mechanical derived force fields (QMD-FFs) in the de novo design of self-assembled materials. Using a QMD-FF based solely on the target molecular structure, we successfully simulated the spontaneous transition to an ordered liquid crystal phase, capturing conformational changes and ensemble reorganization over 200 ns. This multiscale characterization aligns quantitatively with experimental data, bridging various time/length scales and marking a significant advancement in the bottom-up design of supramolecular materials.
Publications
M. Ortega, J. G. Vilhena and R. Pérez. “Hygroscopy of Single‐Stranded DNA Nano‐Brushes: Atomic Workings of its Hydration‐Induced Deformation“. Advanced Materials Interfaces 9 (34), 2201272 (2022).
L. G. da Silveira, P. R. Livotto, D. Padula, J. G. Vilhena, and G. Prampolini. “Predicting Spontaneous Orientational Self-Assembly: In Silico Design of Materials with Quantum Mechanically Derived Force Fields”. The Journal of Physical Chemistry Letters 13 (1), 243 (2022) — featured in the front cover.
S. Scherb, A. Hinaut, R. Pawlak, J. G. Vilhena, Y. Liu, S. Freund, Z. Liu, X. Feng, K. Müllen, T. Glatzel, A. Narita, Ernst Meyer. “Giant thermal expansion of a two-dimensional supramolecular network triggered by alkyl chain motion“. Communications Materials 1, 8 (2020).
