Method Development
Pushing the limits of all-atom simulations
All-atom molecular dynamics (MD) simulations are arguably one of the most widely used methods to unveil atomic processes at the nanoscale. From the spontaneous self-assembly of supramolecular networks to deciphering the mechanical properties controlling DNA activity inside cells, MD has successfully delivered atomic-level insights into a wide range of processes. However, its reliability hinges on the accuracy of the adopted force-fields (FFs).
While general force-fields often adequately account for the structure and dynamics of large biomolecules, they frequently fall short for supramolecular materials. The structure and dynamics of these materials are governed by a delicate balance between weak intermolecular interactions and flexible internal degrees of freedom, which general FFs often misrepresent. A paradigmatic example are liquid crystals, where standard FFs overestimate transition temperatures by more than 50K. Although several remedies have been proposed, they often compromise predictive power by relying on empirical data, thus hampering in-silico material design.
In a collaborative effort lead by the Prof. Giacomo Prampolini (CNR Pisa), in the past years, we have developed an automated method to obtain all force-field (FF) parameters, both bonded and non-bonded, for arbitrarily large molecules solely from ab-initio quantum mechanical data. This approach, which is the first of its kind, significantly outperforms commonly used general force-fields, as evidenced by its agreement with experimental data. Our methods offers both first-principles-based predictivity and unprecedented accuracy, which are the most desired attributes for in-silico materials design.
Publications
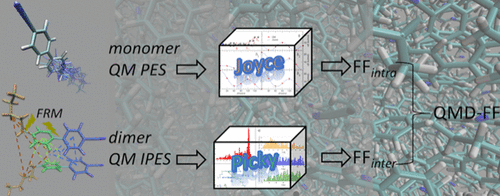
S. Giannini, P. M. Martínez, A. Semmeq, J. P. Galvez, A. Piras, A. Landi, D. Padula, J. G. Vilhena, J. Cerezo and G. Prampolini. “JOYCE3.0: A General Protocol for the Specific Parametrization of Accurate Intramolecular Quantum Mechanically Derived Force Fields“. J. Chem. Theory Comput. 6 (21), 3156-3175 (2025).

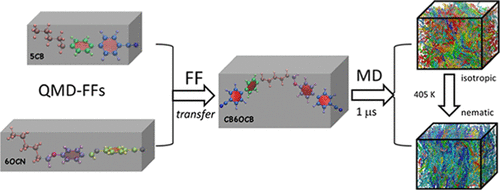
J. G. Vilhena, L. G. da Silveira, P. R. Livotto, I. Cacelli and G. Prampolini. “Accurate Quantum-Mechanically Derived Force-Fields through a Fragment-Based Approach: Balancing Specificity and Transferability in the Prediction of Self-Assembly in Soft Matter”. Journal of Chemical Theory and Computation 18 (11), 6905-6919 (2022).
J. G. Vilhena, L. G. da Silveira, P. R. Livotto, I. Cacelli and G. Prampolini. “Automated Parameterization of Quantum Mechanically Derived Force-Fields for Soft Materials and Complex Fluids: development and validation“. Journal of Chemical Theory and Computation 17 (7), 4449 (2021).
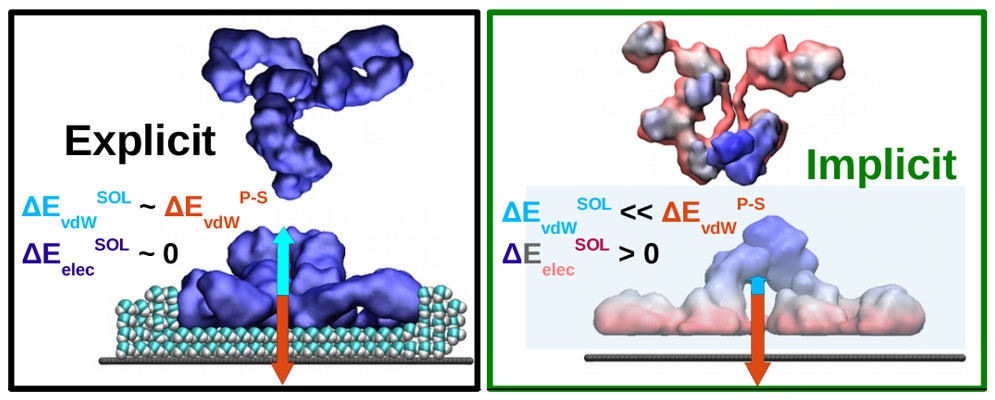
M. Ortega, J. G. Vilhena, P. Rubio, P. A. Serena and R. Pérez. “Assessing the accuracy of implicit solvation models to describe the IgG adsorption over a hydrophobic surface“. Journal of Chemical Theory and Computation 15 (4), 2548 (2019).